







Cập nhật di truyền học trong chẩn đoán và điều trị rối loạn phát triển thần kinh
HỘI NGHỊ KHOA HỌC TÂM THẦN HỌC TOÀN QUỐC LẦN THỨ V
Báo cáo viên: Ths. Lê Công Thiện
Đơn vị: Bộ môn Tâm thần – Trường Đại học Y Hà Nội
Viện Sức khỏe Tâm thần – Bệnh viện Bạch Mai
Khái niệm:
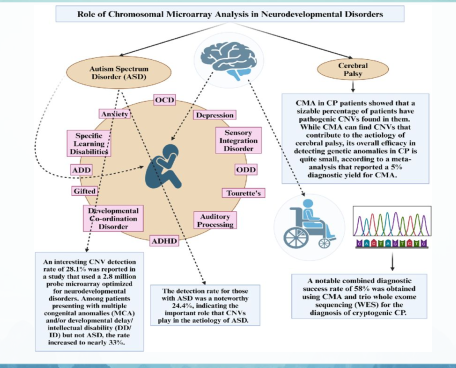
- Rối loạn phát triển thần kinh (Neurodevelopmental Disorders, NDD) là những bệnh mạn tính phổ biến nhất trong chăm sóc sức khỏe ban đầu cho trẻ em.
- Theo DSM-5, NDD bao gồm khuyết tật trí tuệ (ID), chậm phát triển toàn diện (GDD), rối loạn giao tiếp (rối loạn ngôn ngữ, rối loạn âm thanh lời nói, rối loạn nói lắp khởi phát ở trẻ em và rối loạn giao tiếp xã hội), rối loạn phổ tự kỷ (ASD), rối loạn tăng động giảm chú ý (ADHD), rối loạn học tập đặc hiệu (liên quan đến đọc, diễn đạt bằng văn bản và/hoặc toán học) và rối loạn vận động (rối loạn phối hợp phát triển, rối loạn vận động rập khuôn và rối loạn tic).
- Khái niệm rộng hơn về NDD bao gồm các tình trạng nằm ngoài phạm vi của DSM-5 như bại não (CP), động kinh, và đôi khi là các rối loạn tâm thần mà có bằng chứng lâm sàng và sinh học mạnh mẽ về nguồn gốc phát triển như tâm thần phân liệt.
- NDD được đặc trưng bởi sự thiếu hụt phát triển về nhận thức, ngôn ngữ, hành vi và/hoặc kỹ năng vận động gây ra suy giảm chức năng cá nhân, xã hội, học tập và/hoặc nghề nghiệpk
Dịch tễ
- Theo dữ liệu từ nghiên cứu Gánh nặng Bệnh tật Toàn cầu (GBD), chỉ tính riêng ở nhóm trẻ emvà trẻ vị thành niên (từ 0–14 tuổi), tổng số ca mắc của 3 dạng NDD phổ biến nhất (Rối loạn phổtự kỷ - ASD, Rối loạn tăng động giảm chú ý - ADHD, và Khuyết tật trí tuệ - ID) đạt khoảng 235 triệuca.
- Theo GBD, tỷ lệ ADHD chiếm khoảng 1,1% dân số toàn cầu. Nếu xét riêng trong quần thể trẻ em và vị thành niên, tỷ lệ này dao động trung bình 5-7%.
- ID chiếm khoảng 0,9-1,16%, chủ yếu tập trung tại các quốc gia có chỉ số phát triển kinh tế - xã hội (SDI) từ thấp đến trung bình.
- ASD chiếm khoảng 0,6% đến 0,79% dân số toàn cầu. WHO công bố ước tính trung bình là 1 trên 127 người (khoảng 0,78%) mắc ASD. Ở các quốc gia phát triển có hệ thống sàng lọc tốt
- như Hoa Kỳ, tỷ lệ được báo cáo ở trẻ em hiện tại đã lên tới 1 trên 36 trẻ (~2,8%).
- Nam giới chiếm tỷ lệ cao rõ rệt ở các NDD. Tỷ lệ nam/nữ ở ASD là 4/1.
- Hiện tượng đồng mắc nhiều rối loạn NDD trên cùng một cá thể là phổ biến hơn mắc đơn độc một loại rối loạn NDD.k to edit Master titl
Nguyên nhân di truyền học
- Nguyên nhân gây ra NDD rất phức tạp bao gồm cả yếu tố di truyền và môi trường học.
- Năm 1977, một nghiên cứu về cặp song sinh do Rutter và Folstein thực hiện cho thấy tỷ lệ tương đồng đối với ASD cao hơn đáng kể ở cặp song sinh cùng trứng so với cặp song sinh khác trứng 🡪 các yếu tố di truyền có thể đóng vai trò quan trọng trong ASD, tính di truyền xấp xỉ 60–90%.
- Trong thập kỷ qua, sự tiến bộ của công nghệ giải trình tự gen cao, đặc biệt việc giải trình tự toàn bộ exome (WES) và giải trình tự toàn bộ bộ gen (WGS) quy mô lớn đã thúc đẩy mạnh mẽ những phát hiện về di truyền của NDD.
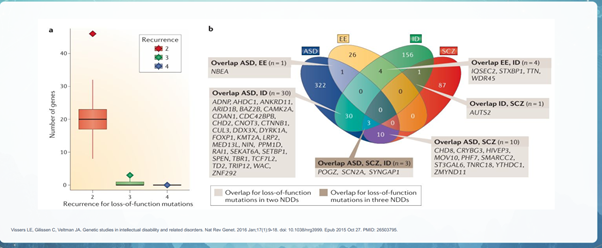
- Nhiều dạng biến thể khác nhau đã được báo cáo là góp phần gây ra NDD, như đột biến de novo (DNM), biến thể số lượng bản sao (CNV), biến thể di truyền hiếm gặp (RIV) và biến thể phổ biến. Cho đến nay đã xác định được hơn 200 gen và vị trí có nguy cơ cao, tuy nhiên chỉ khoảng 30% trường hợp NDD có thể được quy cho nguy cơ di truyền đã biết và vẫn còn nhiều yếu tố di truyền khác cần được xác định.
- Trong tương lai, nghiên cứu di truyền vẫn rất quan trọng để giải mã đầy đủ căn nguyên và tính đa dạng của NDD
Đột biến de novo (DNM)
- Đột biến de novo là những đột biến ngẫu nhiên không di truyền phát sinh trong tế bào mầm hoặc trong quá trình phát triển phôi sớm.
- Các đột biến làm mất chức năng gen (dnLGD) như đột biến dịch khung, đột biến vô nghĩa xuất hiện với tần suất cao gấp đôi ở người mắc tự kỷ so với anh chị em ruột không mắc bệnh của họ.
- Các đột biến sai nghĩa (dnMIS) có xu hướng tập trung tại các vùng "điểm nóng" (hotspots) trên một số gen nhất định, đặc biệt là các gen liên quan đến kênh ion (như GRIN1, GRIN2B, KCNK3, KCNQ2)
Các biến thể di truyền hiếm gặp (Rareinherited variants-RIV)
- Các biến thể hiếm gặp trong NDDs thường có tần suất xuất hiện cực thấp trong quần thể chung (thường dưới 0.1% hoặc chỉ xuất hiện ở dạng đơn độc trong một gia đình), nhưng lại mang tínhthấm (penetrance) rất cao, nghĩa là nếu cá thể sở hữu biến thể đó thì nguy cơ biểu hiện bệnh làcực kỳ lớn.
- Chủ yếu tồn tại dưới 2 dạng: biến thể số lượng bản sao (CNV) và biến thể đơn nucleotide (SNVs) hoặc các đột biến chèn/mất đoạn nhỏ (Indels).
- 2 đặc tính: tính đa hình (Cùng một biến thể hiếm gặp có thể gây ra các kiểu hình lâm sàng hoàn toàn khác nhau ở những cá thể khác nhau) và dị hợp (Nhiều biến thể hiếm gặp ở các gen khác nhau có thể dẫn đến cùng một hội chứng lâm sàng hoặc hành vi giống hệt nhau, do chúng cùng tấn công vào một con đường tín hiệu sinh học)
Biến thể số lượng bản sao (Copy number variations-CNV)
- CNV là dạng đột biến cấu trúc, mất đi hoặc nhân lên trình tự DNA có kích thước đáng kể từ kilobase (Kb) đến megabase (Mb), thường trải rộng trên nhiều gen khác nhau.
- Tỷ lệ CNV lớn (>250 kb) cao hơn mức bình thường ở những người mắc NDD so với nhóm đối chứng đã được xác nhận.
- Các đột biến mất đoạn có tác động lớn hơn so với đột biến lặp đoạn.
- Một số vị trí CNV tái phát liên quan đến NDD đã được xác định như 1q21.1, 3q29, 7q11.23,15q11.2, 16p11.2 và 22q11.2, với ước tính cho thấy 20% bệnh nhân mắc ASD mang ít nhất một CNV.
- Trong đó, 16p11.2 là vị trí nguy cơ được nghiên cứu nhiều nhất của NDD. Cả việc mất và nhân đôi 16p11.2 đều có thể dẫn đến các kiểu hình liên quan đến NDD, bao gồm suy giảm xã hội, hành vi lặp đi lặp lại, chậm phát triển ngôn ngữ và khuyết tật trí tuệ
Các biến thể phổ biến
- Trái ngược với các đột biến hiếm gặp, các biến thể di truyền phổ biến – chủ yếu là đa hình đơn nucleotide (SNPs) có tần suất alen trong quần thể lớn hơn 1% – sở hữu mức độ tác động lâm sàng của từng biến thể riêng lẻ cực kỳ nhỏ.
- Tuy nhiên, khi hàng nghìn biến thể phổ biến này tích tụ và hiệp đồng tác động trên cùng một cá thể thì tạo nên một nền tảng nguy cơ di truyền đa gene (polygenic background) vững chắc, đóng góp từ 10% đến hơn 50% khả năng di truyền (heritability) của các NDDs.
- SNPs phổ biến trong quần thể đóng góp đến gần 50% khả năng di truyền của tự kỷ (thông qua các nghiên cứu
- GWAS). Các nghiên cứu di truyền học quần thể mới nhất chỉ ra rằng điểm nguy cơ đa gen (PRS) cao về các tính trạng nhận thức và tâm thần được di truyền trực tiếp từ cha mẹ không bị bệnh. Nền tảng đa gen này có thể tương tác trực tiếp bằng cách làm trầm trọng thêm kiểu hình của một đột biến mới xuất hiện (de novo), hoặc tích tụ độc lập để đẩy đứa trẻ vượt qua ngưỡng dung nạp của não bộ dẫn đến biểu hiện bệnh.
- Các biến thể phổ biến này thường thể hiện tính đa hiệu di truyền rất cao, cho thấy sự chồng lấp và tương quan di truyền mạnh mẽ giữa các NDDs với nhau cũng như với các rối loạn tâm thần khởi phát ở tuổi trưởng thành.
- Sự hội tụ di truyền của nhóm biến thể phổ biến này chủ yếu khu trú ở các vùng gen điều hòa biểu hiện hoạt động mạnh trong giai đoạn phát triển não bộ phôi thai, ảnh hưởng đến các con đường tín hiệu điều hòa tính mềm dẻo của synap và sự biệt hóa tế bào thần kinh
Mô hình di truyền của NDD
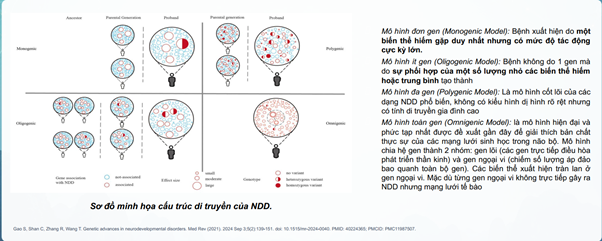
Các quá trình bị ảnh hưởng trong NDD ở não bộđang phát triển và não bộ trưởng thành
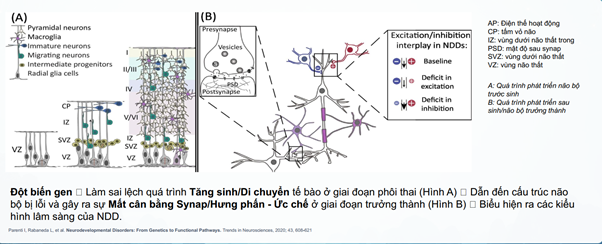
Sự chồng chéo gen ở các rối loạn phát triển
- Di truyền biểu sinh (epigenetics) đóng vai trò như một bộ điều khiển phân tử, định hướng cách tế bào đọc mã di truyền mà không làm thay đổi trình tự nucleotit gốc của DNA.
- Quá trình này được duy trì qua 3 con đường cốt lõi:
- sự methyl hóa DNA tại các đảo CpG
- sự biến đổi hóa học ở đuôi histone (như acetyl hóa hoặc methyl hóa) làm thay đổi cấu trúc không gian của chromatin
- sự tinh chỉnh sau phiên mã của các RNA không mã hóa (ncRNAs).
- Khi các cơ chế này bị đảo lộn — do đột biến de novo ở các gene mã hóa cho các enzyme biểu sinh hoặc do tác động tiêu cực từ môi trường ngoại sinh — cấu trúc chromatin sẽ bị biến đổi 🡪 sự sai lệch trong quá trình biệt hóa tế bào gốc thần kinh, sự di chuyển của neuron và định hình tính mềm dẻo của synap
🡪 Hệ quả của những rối loạn điều hòa biểu sinh này là sự khởi phát của NDD.
Cơ chế di truyền biểu sinh
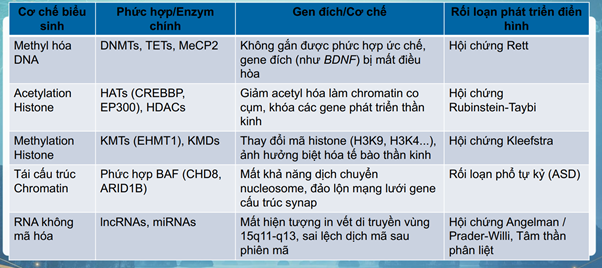
Tiến bộ trong Chẩn đoán Di truyền
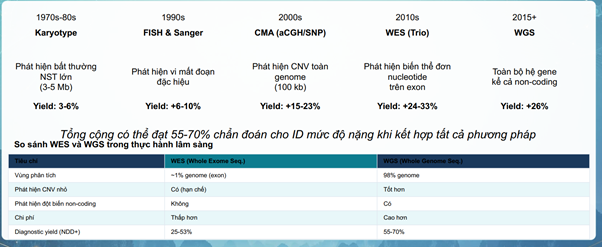
Karyotype (Nhiễm sắc thể đồ)
- Karyotype là kỹ thuật khảo sát cấu trúc và số lượng của toàn bộ bộ nhiễm sắc thể (NST) trong tế bào ở kỳ giữa(metaphase) của quá trình phân bào – thời điểm các NST co xoắn cực đại và nhìn rõ nhất dưới kính hiển vi quang học.
- Karyotype giúp phát hiện các bất thuờng về số lượng NST như hội chứng Down, hội chứng Patau, Klinefelter, Turner hay các tái cấu trúc NST lớn như chuyển đoạn cân bằng và không cân bằng liên quan đến nguy cơ tái phát ở thế hệ sau hoặc đảo đoạn, mất đoạn lớn NST.
- Tuy nhiên phần lớn nguyên nhân gây NDD nằm ở cấp độ vi mất đoạn/vi lặp đoạn có kích thuớc dưới 5Mb hoặc các đột biến điểm nằm sâu trong exon mà Karyotype không phát hiện được.
FISH
- FISH là kỹ thuật di truyền tế bào phân tử sử dụng các đoạn mồi DNA ngắn(probe) được gắn tín hiệu huỳnh quang để tìm và liên kết chính xác vào các vùng trình tự DNA tương đồng trên nhiễm sắc thể của người bệnh.
- Độ phân giải: Đạt mức 100 Kilobases (Kb) đến 1 Megabase (Mb). Độ phân giải này cao hơn gấp 10 lần so với xét nghiệm Karyotype truyền thống, cho phép
- FISH nhìn thấy được các tổn thương vi mô. FISH có thể chẩn đoán xác định các hội chứng vi mất đoạn, vi lặp đoạn mà
- Karyotype thuờng bỏ sót như Hội chứng DiGeorge (mất đoạn 22q11.2), hội chứng Williams (mất đoạn 7q11.23), hội chứng Angelman / Prader-Willi (mất đoạn 15q11-q13).
- Hạn chế: FISH là xét nghiệm định hướng. Nghĩa là bác sĩ phải có nghi ngờ lâm sàng rất rõ ràng về một hội chứng cụ thể để chọn đúng loại đoạn mồi huỳnh quang (probe) tương ứng. Nếu chọn sai đích, FISH sẽ cho kết quả âm tính giả và hoàn toàn không thể phát hiện ra các đột biến nằm ngoài vùng dò

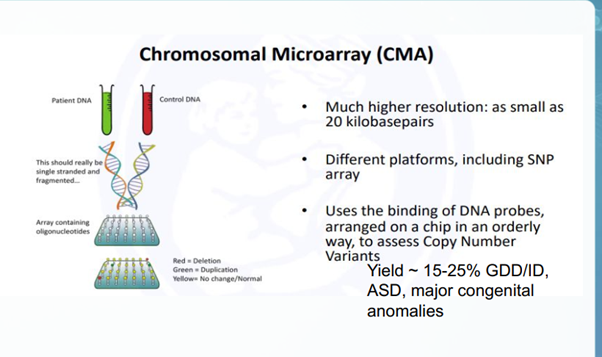
Chromosomal Microarray (CMA)
Nguyên lý: CMA là kỹ thuật quét toàn bộ hệ gen trên một chip sinh học nhằm phát hiện sự mất cân bằng về mặt số lượng của các đoạn DNA. Xét nghiệm này bao gồm hai loại chính: array-CGH (lai so sánh hệ gen) và SNP-array (phân tích đa hình đơn nucleotide).
Mục đích: tìm các Biến thể số lượng bản sao (CNV), tức là các vùng vi mất đoạn (microdeletions) hoặc vi lặp đoạn (microduplications) kích thước siêu nhỏ dưới ngưỡng nhìn của kính hiển vi.
Độ phân giải: Đạt mức cực cao từ 20 Kb đến 100 Kb(cao hơn Karyotype truyền thống gấp 100 lần).
CMA là Xét nghiệm di truyền đầu tay cho 3 nhóm đối tượng: Chậm phát triển trí tuệ/chậm phát triển tâm vận động (DD/ID), Rối loạn phổ tự kỷ (ASD) và Đa dị tật bẩm sinh không rõ nguyên nhân.
Chromosomal Microarray (CMA)
CMA có thể phát hiện
- Sự tăng/giảm biến thể số lượng bản sao (CNV),
- Vùng đồng hợp tử CMA hạn chế phát hiện
CMA hạn chế phát hiện
- Trình tự DNA lặp lại bao gồm sự mở rộng lặp lại trinucleotide (ví dụ: sự mở rộng lặp lại FMR1 )
- Sự sắp xếp lại nhiễm sắc thể cân bằng
- Biến thể ở cấp độ trình tự trong exome/genome
- Biến thể ty thể
- Thay đổi biểu sinh (ví dụ: bất thường về methyl hóa, dị hợp tử đơn bội)
- Thể khảm ở mức độ thấp
Hiệu quả chẩn đoán của CMA trong thực hành lâm sàng
- Karyotype thông thường chỉ tìm ra nguyên nhân ở khoảng 3% bệnh nhân NDD. Trong khi đó, CMA đẩy tỷ lệ chẩn đoán thành công lên tới 15% – 25% nhờ việc phát hiện ra hàng loạt hội chứng vi mất/lặp đoạn phổ biến
- CMA không chỉ báo có lỗi hay không, mà còn xác định chính xác tọa độ đứt gãy (breakpoints) và liệt kê cụ thể các gene nằm trong vùng bị mất hoặc lặp đó, giúp bác sĩ tiên lượng kiểu hình lâm sàng của trẻ.
- Phát hiện vùng đồng hợp tử lớn (chỉ có ở SNP-array): SNP-array có thể phát hiện các vùng mất tính dị hợp tử (Regions of Homozygosity - ROH) 🡪 gợi ý tình trạng đồng huyết thống hoặc lệch nhiễm sắc thể từmột phía (Uniparental Disomy - UPD) – nguyên nhân gây ra hội chứng Prader-Willi hoặc Angelman.
- Các nghiên cứu chỉ ra kết quả CMA làm thay đổi chiến lược quản lý điều trị (như chụp chiếu tầm soát cơ quan đích, can thiệp tâm lý sớm, theo dõi động kinh đi kèm) ở hơn 70% bệnh nhân có kết quả bất thường.
Whole Exome Sequencing (WES)
WES
Nguyên lý: WES sử dụng công nghệ giải trình tự thế hệ mới (NGS) để đọc trình tự
của toàn bộ Exome — gồm tất cả các vùng mã hóa protein (exons) của khoảng 20.000 gen trong cơ thể con người.
Mục đích: tìm các Biến thể đơn nucleotide (SNVs)và các đột biến chèn/mất đoạn nhỏ (Indels) nằm sâu bên trong cấu trúc gene
Độ phân giải: Đạt mức tối đa ở cấp độ từng nucleotide đơn lẻ(độ phân giải cao nhất trong các xét nghiệm gen đích hiện tại). Mặc dù exome chỉ chiếm khoảng 1.5% toàn bộ hệ gen, nhưng nó lại chứa tới 85%các đột biến gây bệnh đã biết ở người.
Ưu điểm
- Độ bao phủ vùng mã hóa cực cao: Đọc trình tự của toàn bộ ~ 20.000 gene cùng lúc.
- Độ phân giải tối đa: Phát hiện chính xác các đột biến điểm (SNVs) và chèn/mất đoạn nhỏ (Indels) ở cấp độ từng nucleotide.
- Hiệu suất chẩn đoán vượt trội: Đạt tỷ lệ ra kết quả từ 30% – 43% ở các ca NDD khó, chậm phát triển trí tuệ hoặc tự kỷ thể nặng.
Hạn chế
- Thường bỏ sót các biến thể số lượng bản sao (CNVs), vi mất đoạn/vi lặp đoạn lớn.
- Không phát hiện được đột biến lập đoạnchuỗi nu: (Ví dụ: hội chứng nhiễm sắc thể Xdễ gãy - Fragile X).
- Bỏ sót vùng không mã hóa: Không đọc được đột biến ở vùng Intron hoặc vùng điều hòa (Regulatory regions).
- Thách thức về mặt diễn giải dữ liệu: Dễ ra kết quả "Biến thể chưa rõ ý nghĩa lâm sàng" (VUS - Variants of Uncertain Significance).
- WGS (whole Genome Sequencing – Giải trình tự toàn bộ gen)
- Nguyên lý: WGS là công nghệ giải trình tự thế hệ mới (NGS) cao cấp nhất, cho phép đọc toàn bộ cấu trúc DNA của một cá thể, bao gồm cả vùng mã hóa protein (Exon - chiếm 1.5%) và vùng không mã hóa (Intron/Regulatory regions - chiếm 98.5%).
- Mục đích: Là xét nghiệm "tất cả trong một" (All-in-one). WGS có khả năng phát hiện đồng thời cả đột biến điểm (SNVs), đột biến chèn/mất đoạn nhỏ (Indels), biến thể số lượng bản sao (CNVs - vi mất/lặp đoạn) và các tái cấu trúc nhiễm sắc thể lớn (chuyển đoạn, đảo đoạn).
- Độ phân giải: Đạt mức tối đa ở cấp độ từng nucleotide đơn lẻ trên quy mô toàn hệ gen WGS là xét nghiệm tối ưu nhất nhưng chi phí cao
Tiếp cận đánh giá nguyên nhân ditruyền trong GDD, ID và/hoặc ASD
Bước 1. Khám sức khỏe toàn diện và thu thập tiền sử phát triển, bệnh sử và tiền sử gia đình.
- Nếu nghi ngờ nguyên nhân cụ thể 🡪 xét nghiệm di truyền chuyên biệt phù hợp (ví dụ: giải trình tự PTEN và phân tích mất/nhân đôi, giải trình tự MECP2 và phân tích mất/nhân đôi, phân tích lặp lại CGG FMR1 ).
- Nếu không nghi ngờ nguyên nhân cụ thể 🡪 bước 2.
Bước 2. Tiến hành kiểm tra tổng quát các biến thể trình tự exon gây bệnh và biến thể số lượng bản sao.
- Cách hiệu quả nhất để thực hiện điều này là thông qua giải trình tự exon (ES) với việc xác định biến thể số lượng bản sao (CNV).
- Nên tiến hành ES như là đánh giá ban đầu khi có thể.
- Nên thực hiện ES bằng cách sử dụng phân tích bộ ba khi có thể để giảm số lượng biến thể ứng cử viên và cung cấp thông tin cho việc phân loại biến thể.
- Nếu không xác định được nguyên nhân 🡪 bước 3.
Bước 3. Hoàn thành việc phân tích lại định kỳ các biến thể đã báo cáo và dữ liệu exon; Cân nhắc thực hiện thêm các xét nghiệm di truyền (ví dụ: giải trình tự bộ gen, phân tích lặp lại CGG của gen FMR1 nếu chưa thực hiện, xét nghiệm tế bào học, nếu cần thiết, dựa trên kết quả ES/CMA và các phát hiện lâm sàng) và/hoặc chuyển đến chuyên khoa di truyền y học để đánh giá nếu được chỉ định .
Di truyền học trong điều trị NDD
Thay vì chỉ điều trị triệu chứng hành vi bằng thuốc hướng thần, các liệu pháp mới tập trung sửa chữa tận gốc tổn thương ở cấp độ phân tử và gen.
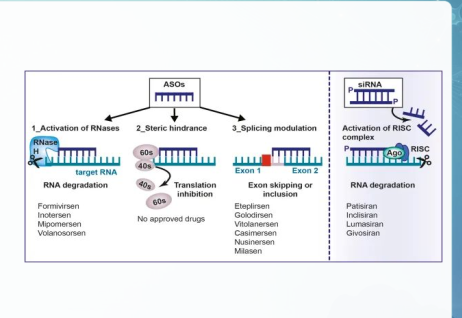
Liệu pháp Antisense Oligonucleotides
Là liệu pháp sử dụng các đoạn nucleotide ngắn, biến đổi cấu trúc để liên kết đặc hiệu với mRNA mục tiêu.
Giúp sửa lỗi dịch mã, phục hồi protein thiếu hụt hoặc bất hoạt RNA độc hại.
❖ Các cơ chế hoạt động chính là:
1) Phân cắt RNA thông qua việc huy động các enzyme nội sinh
2) Cản trở không gian
3) Điều chỉnh quá trình ghép nối
4) Kích hoạt phức hợp RISC bởi siRNA mạch đôi.
Ưu điểm
- Độ chính xác rất cao, tác động đúng gene đích lâm sàng.
- Ít nguy cơ gây đột biến chèn đoạn ngẫu nhiên vào DNA của người bệnh.
- Đã có bằng chứng lâm sàng thành công ở một số NDDs (như Hội chứng Angelman).
Nhược điểm
Không duy trì vĩnh viễn, phải tiêm lặp lạiđịnh kỳ vào tủy sống (intrathecal).
- Giá thành điều trị cực kỳ đắt .
- Khó tiếp cận đồng đều đến toàn bộ các vùng sâu trong não bộ.
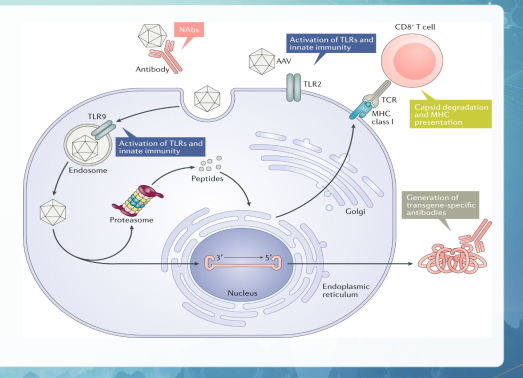
Liệu pháp Thay thế Gen (qua Vector AAV)
Vector virus liên kết với adeno (AAV) là nền tảng hàng đầu cho việc truyền
gen để điều trị nhiều loại bệnh ở người.
Cơ chế: Sử dụng virus đã bất hoạt (phổ biến nhất là AAV9 có khả năng xuyên qua hàng rào máu não) để đưa một bản sao gene lành lặn vào tế bào, thay thế cho gene bị lỗi.
Ưu điểm
Hiệu quả lâu dài, tiềm năng chữa khỏi bệnh chỉ với một lần tiêm duy nhất trong
đời
Phù hợp cho các hội chứng NDD đơn gene do mất chức năng (như Hội chứng Rett, Hội chứng SHANK3).
Nhược điểm
Nguy cơ kích hoạt phản ứng miễn dịchnghiêm trọng chống lại vector virus.
• Giới hạn kích thước gene hình học (AAV không chở được các gene quá lớn).
• Rủi ro độc tính trên gan khi dùng liều cao toàn thân.
Chỉnh sửa Gen trực tiếp
Khác với các liệu pháp can thiệp ở tầng RNA (như ASOs) có tính chất tạm thời, công nghệ chỉnh sửa gen tác động trực tiếp và vĩnh viễn vào DNA cốt lõi trong nhân tế bào thần kinh, sử dụng hệ thống enzyme để cắt, sửa đổi hoặc "thay thế" trực tiếp các đột biến điểm ngay trên chuỗi DNA gốc
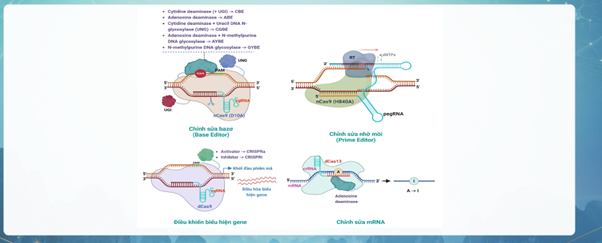
【CRISPR-Cas9】Cắt đứt sợi đôi DNA
Rủi ro: Lỗi chèn/mất đoạn do sửa chữa chủ yếu bằng con đường nối kết đầu tận không tương đồng (NHEJ)
【Base Editing】 (Đổi 1 nucleotide đơn)
An toàn hơn: không cắt DNA
Hạn chế: Chỉ sửa đổi được một số ít loại chuyển đổi base
【Prime Editing】Tìm và thay thế nâng cao
Tối ưu: không cắt DNA, sửa được cả đột biến điểm, chèn và mất đoạn nhỏ
Điều trị Đích bằng Phân tử nhỏ(Targeted Small Molecules)
Targeted Small Molecules không can thiệp vào cấu trúc gen, mà dùng thuốc để điều chỉnh các con đường tín hiệu xuôi dòng (downstream pathways) bị rối loạn do gen đột biến gây ra.
- Mặc dù có hàng trăm gen đột biến hiếm gặp và phổ biến khác nhau gây ra NDD, các protein do chúng mã hóa lại có xu hướng hội tụ (converge) vào một số trục tín hiệu nội bào và cấu trúc synap nhất định. Việc sử dụng các phân tử nhỏ hướng đích nhằm khôi phục lại trạng thái sinh lý bình thường của não bộ.
- Cơ chế thông qua trục tín hiệu PI3K/Akt/mTOR (Điều hòa dịch mã và sinh trưởng tế bào) và hệ thống thụ thể Glutamate (mGluR5 và NMDAR).
Điều trị Đích bằng Phân tử nhỏ(Targeted Small Molecules)
Trục tín hiệu PI3K/Akt/mTOR (Điều hòa dịch mã và sinh trưởng tế bào):
- Đường truyền tín hiệu mTOR (mammalian target of rapamycin) là trung tâm điều hòa quá trình tổng hợp protein tại synap, sự phát triển đuôi gai và tính mềm dẻo của synap.
- Trong nhiều thể NDDs đơn gen (như Xơ cứng củ - TSC do đột biến TSC1/TSC2, Hội chứng dị dạng mạch não do đột biến PTEN), trục mTOR bị kích hoạt quá mức (hyperactivation), dẫn đến việc tổng hợp protein mất kiểm soát, làm sai lệch cấu trúc khớp thần kinh.
- Các phân tử nhỏ hoạt động như chất ức chế kinase sẽ ức chế sự gắn phosphate, đưa hoạt tính mTOR trở lại ngưỡng cân bằng.
Hệ thống thụ thể Glutamate (mGluR5 và NMDAR):
- Glutamate là chất dẫn truyền thần kinh hưng phấn chính trong não bộ.
- Trong Hội chứng Fragile X (FXS) (do mất protein FMRP), trục tín hiệu qua thụ thể metabotropic glutamate receptor 5 (mGluR5) bị phóng đại, gây tăng tổng hợp protein synap dị biệt và tăng hiện tượng sụt giảm synap kéo dài. Các phân tử nhỏ đóng vai trò là chất điều hòa dị lập thể âm tính (NAMs) của mGluR5 giúp kìm hãm trực tiếp trạng thái kích thích này.
- Ngược lại, trong một số thể tự kỷ và chậm phát triển trí tuệ do đột biến các gen GRIN1, GRIN2A, GRIN2B, hoạt tính của thụ thể NMDA (NMDAR) lại bị suy giảm trầm trọng. Các phân tử nhỏ đóng vai trò là chất điều hòa dị lập thể dương tính (PAMs) sẽ gắn vào thụ thể để tăng cường dòng ion canxi đi vào tế bào, khôi phục lại khả năng dẫn truyền.
Ưu điểm
- Sử dụng đường uống hoặc tiêm thông thường, dễ phân bố khắp não bộ và vượt qua hàng rào máu não
- Có thể dùng các thuốc sẵn có (Tái mục đích thuốc - ví dụ: dùng chất ức chế mTOR như Everolimus cho hội chứng Xơ cứng củ)
Nhược điểm
Không sửa chữa được tận gốc nguyên nhân di truyền.
- Phải dùng thuốc suốt đời.
- Thường có nhiều tác dụng phụ toàn thân do thuốc tác động lên các mô cơ quan khác ngoài não
Người đưa tin: Linh Hoa













